In Vivo Gene Cloning (AQA A-Level Biology): Revision Notes
In Vivo Gene Cloning
In vivo gene cloning is the process of producing copies of a specific gene within living organisms. After DNA has been cut into fragments using restriction endonucleases, scientists need to identify and clone the fragment containing the desired gene. This can be achieved through two main approaches:
- In vivo cloning: transferring DNA fragments to host cells using vectors
- In vitro cloning: using polymerase chain reaction (PCR)
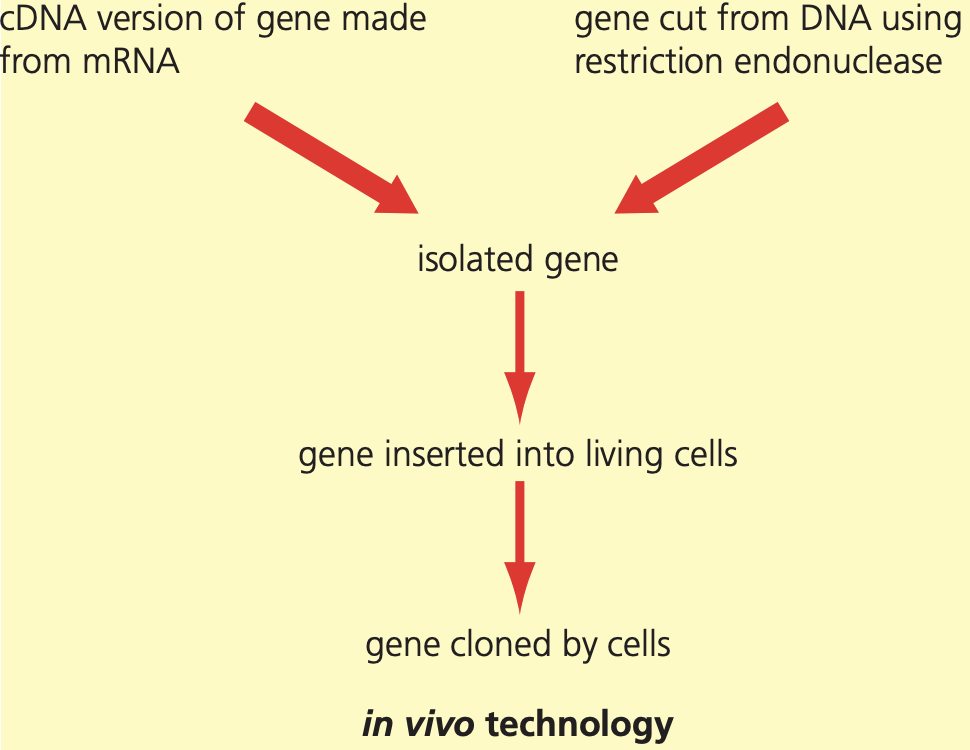
This technique allows scientists to produce sufficient quantities of genes for medical or commercial applications, making it a cornerstone of biotechnology and genetic engineering.
Importance of sticky ends
When restriction endonucleases cut DNA, they create specific cutting patterns at sequences called recognition sites. These enzymes often cut DNA in a staggered fashion, leaving short single-stranded overhangs known as sticky ends.
The nucleotides on these single-stranded overhangs are complementary to those on the opposite side of the cut, since they were previously base-paired in the double helix. This complementary nature is what makes the ends "sticky" - they can readily bind to other DNA fragments cut with the same restriction enzyme.
When fragments with complementary sticky ends come together, the enzyme DNA ligase joins them permanently by forming bonds between the phosphate-sugar backbone of the DNA strands. This process creates recombinant DNA - DNA that combines genetic material from different sources.
The significance of sticky ends lies in their ability to join DNA from any organism, provided the same restriction endonuclease was used to cut both pieces. This universal compatibility makes genetic engineering possible across species boundaries.
Preparing the DNA fragment for insertion
Before a DNA fragment can function properly in a host cell, additional DNA sequences must be attached to enable gene expression.
Promoter regions
For transcription to occur, RNA polymerase must attach to a specific binding site called a promoter. This region contains the necessary sequences that both RNA polymerase and transcription factors recognise to initiate mRNA synthesis. Without an appropriate promoter, the inserted gene cannot be transcribed.
Terminator regions
At the other end of the DNA fragment, a terminator region must be added. This sequence signals RNA polymerase to release from the DNA and stop transcription at the correct point. Terminator regions ensure that transcription produces the right length of mRNA.
Both promoter and terminator sequences are essential regulatory elements that control when, where, and how much protein is produced from the inserted gene. Without these sequences, even successfully inserted genes will not function properly.
Insertion of DNA fragment into a vector
Once the DNA fragment has been prepared with promoter and terminator regions, it must be inserted into a vector - a DNA molecule that can transport the fragment into host cells.
Plasmids as vectors
The most commonly used vectors are plasmids - small, circular pieces of DNA found naturally in bacteria. Plasmids exist separately from the main bacterial chromosome and can replicate independently. Most plasmids carry genes for antibiotic resistance, which makes them useful for identifying successful transformations.
The insertion process
The same restriction endonuclease used to cut the DNA fragment is also used to open the plasmid. This ensures that both the fragment and the opened plasmid have complementary sticky ends. When the DNA fragment and plasmid are mixed together, some fragments become incorporated into the plasmids through base-pairing of their sticky ends.
Worked Example: Vector Insertion Process
Step 1: Cut the plasmid with the same restriction enzyme used on the DNA fragment
- This creates complementary sticky ends
Step 2: Mix the DNA fragment with the opened plasmid
- Sticky ends base-pair through complementary nucleotides
Step 3: Add DNA ligase
- Forms permanent bonds in the phosphate-sugar backbone
- Creates recombinant plasmids containing the foreign gene
DNA ligase then makes these joins permanent, creating plasmids that now contain the recombinant DNA. These modified plasmids can carry the foreign gene into bacterial cells.
Introduction of DNA into host cells
The modified plasmids must be introduced into bacterial cells through a process called transformation. This involves mixing plasmids and bacterial cells in a medium containing calcium ions.
The transformation process
The calcium ions and controlled temperature changes make the bacterial cell membrane temporarily permeable, allowing plasmids to pass through into the cytoplasm. However, transformation is quite inefficient for several reasons:
- Only about 1% of bacterial cells actually take up plasmids
- Some plasmids close up again without incorporating the DNA fragment
- Sometimes DNA fragment ends join together to form their own separate plasmid
Critical Limitation of Transformation
Since transformation is inefficient, scientists need methods to identify which bacterial cells have successfully taken up plasmids containing the desired gene. This is where marker genes become essential - without them, it would be impossible to distinguish successful transformants from the majority of cells that failed to take up the desired DNA.
Identifying successful transformants
Since transformation is inefficient, scientists need methods to identify which bacterial cells have successfully taken up plasmids containing the desired gene. This is where marker genes become essential.
Marker genes
Marker genes are additional genes placed on vectors that produce easily identifiable characteristics. These genes help scientists distinguish between cells that have taken up the desired DNA and those that have not.
Antibiotic-resistance markers
Many plasmids naturally carry genes for antibiotic resistance. Scientists can exploit these genes as markers by using the following strategy:
Worked Example: Antibiotic Selection Process
Step 1: Grow all bacterial cells on medium containing the antibiotic (e.g., ampicillin)
Step 2: Observe results:
- Cells that took up plasmids will survive because they have acquired resistance genes
- Cells without plasmids will die because they cannot resist the antibiotic
Step 3: Identify surviving colonies as containing the plasmid
However, some cells may take up plasmids but close them without incorporating the new gene. To identify these cells, scientists use replica plating - a technique that uses a second antibiotic-resistance gene that gets disrupted when the desired gene is inserted.
Fluorescent markers
A more modern approach uses genes that code for fluorescent proteins, such as green fluorescent protein (GFP) from jellyfish. When the desired gene is successfully inserted, it disrupts the fluorescent gene, so:
Worked Example: Fluorescent Marker System
Setup: Insert the desired gene into the middle of a fluorescent protein gene
Results:
- Successful transformants do not fluoresce (fluorescent gene is disrupted)
- Failed transformants continue to produce fluorescent protein and glow under UV light
Advantage: Results can be observed immediately under a microscope without waiting for cell growth
Enzyme markers
Another marker system uses genes that produce enzymes like lactase. The enzyme converts a colourless substrate into a blue product. When the desired gene is inserted correctly:
- Successful transformants cannot produce the enzyme and remain colourless
- Failed transformants produce the enzyme and turn blue on the substrate
This creates a simple visual method for identifying successful gene insertion through colour changes that are easily observed.
Growth and cloning
Once bacterial cells containing the desired gene have been identified, they can be cultured on a large scale to produce significant quantities of the protein product. This represents the final stage of in vivo gene cloning - from initial DNA cutting through to mass production of the desired protein.
The entire process demonstrates how molecular biology techniques can be combined systematically to transfer genes between organisms and harness cellular machinery for practical applications.
Key Points to Remember:
- Sticky ends created by restriction endonucleases allow DNA from different sources to be joined together through complementary base-pairing
- DNA fragments must have promoter and terminator regions added before insertion to enable proper gene expression in host cells
- Transformation is inefficient, so marker genes are essential for identifying which bacterial cells have successfully taken up the desired DNA
- Multiple marker systems exist including antibiotic resistance, fluorescent proteins, and enzyme production - each with different advantages for identifying successful transformants
- In vivo gene cloning enables mass production of proteins by harnessing bacterial cellular machinery after successful gene transfer