Techniques in DNA Analysis (OCR A-Level Biology A): Revision Notes
Techniques in DNA Analysis
Introduction to genome structure
Before exploring DNA analysis techniques, it's important to understand how DNA is organized in eukaryotic cells. The genome refers to the complete set of genetic information in an organism, including all DNA sequences in the nucleus and organelles.
Organization of nuclear DNA
Nuclear DNA in eukaryotes is structured into distinct functional regions:
Structural genes contain the instructions for building polypeptides (protein chains). These genes are not continuous sequences - they consist of:
- Exons: coding sequences that will be translated into amino acids
- Introns: non-coding sequences that are removed during mRNA processing
Worked Example: Calculating Polypeptide Length
The gene coding for the β polypeptide of human haemoglobin is base pairs long but contains only three exons totalling bp.
Using the triplet code ( bases per amino acid):
This demonstrates how a large gene can code for a relatively small polypeptide due to the presence of introns.
Regulatory sequences control gene expression:
- Promoters are located upstream of genes and serve as binding sites for RNA polymerase
- Some regulatory genes code for transcription factors (proteins that control transcription)
- Others code for various forms of RNA that regulate gene expression
Between genes are long stretches of DNA previously called "junk DNA", though these regions are now understood to play roles in gene control. This highlights how our understanding of genome organization continues to evolve.
Organellar genomes
In addition to nuclear DNA, eukaryotic cells contain DNA in organelles:
- Mitochondrial DNA (mtDNA) is found in all eukaryotes
- Chloroplast DNA (ctDNA) is present in plants and some protoctists
These organelles originated from prokaryotic organisms and retain prokaryote-like genomes with minimal non-coding DNA.
Polymerase chain reaction
The polymerase chain reaction (PCR) is an automated process that amplifies selected regions of DNA using alternate stages of polynucleotide separation (denaturation) and DNA synthesis (polymerisation) catalysed by DNA polymerase.
PCR is essential when DNA samples are too small for analysis, such as trace amounts of blood at a crime scene. It can produce billions of copies of a specific DNA sequence in just a few hours.
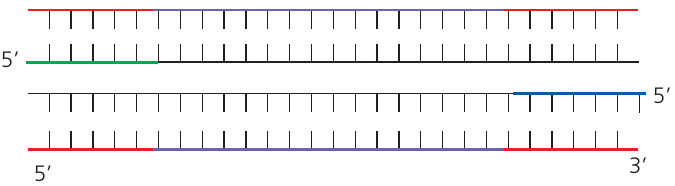
The three stages of PCR
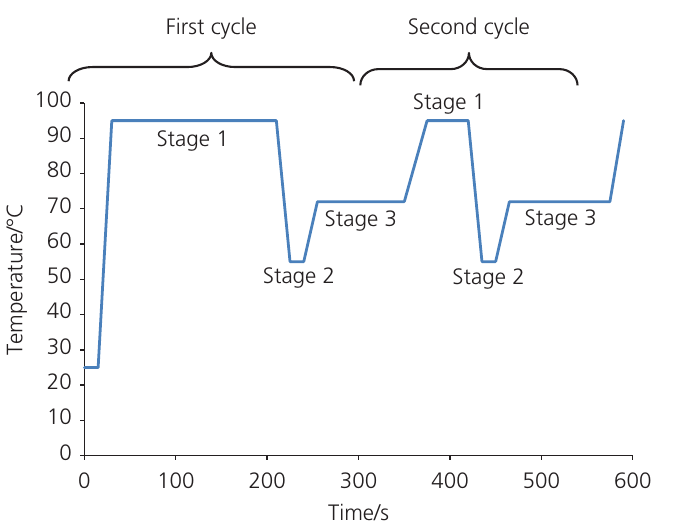
Each PCR cycle lasts approximately minutes and consists of three temperature-controlled stages:
Stage 1: Denaturation
During denaturation, the reaction mixture is heated to 94-95°C. This high temperature breaks the hydrogen bonds between complementary base pairs, causing the double helix to separate into two single-stranded DNA templates.
In the first cycle, denaturation typically lasts about minutes to ensure complete separation. In subsequent cycles, heating for only seconds is sufficient because the DNA fragments being separated are much shorter.
Stage 2: Annealing
The temperature is lowered to 50-65°C to allow primers to bind to the template DNA. Primers are short sequences of DNA (oligonucleotides, typically around nucleotides long) that are complementary to specific regions flanking the target sequence.
Two different primers are used in PCR:
- One primer binds near the end of one template strand
- The other primer binds near the end of the complementary strand
Because DNA strands are antiparallel, the primers attach at opposite ends of the target region. The primer sequences are carefully designed to target a unique location in the genome, ensuring only the desired DNA region is amplified.
Primers serve two critical functions:
- They identify where DNA synthesis should begin
- They provide the double-stranded region needed by DNA polymerase to start adding nucleotides
The high concentration of primers ensures they bind to template strands before the strands can re-anneal to each other.
Primers can be tagged with fluorescent markers to monitor PCR progress and analyse the DNA.
Stage 3: Extension (elongation)
The temperature is increased to 72°C for approximately seconds. At this temperature, Taq polymerase builds new DNA strands complementary to the templates.
The enzyme uses deoxynucleotide triphosphates (dNTPs) as building blocks, adding nucleotides to the end of each primer. DNA synthesis proceeds in the 5' → 3' direction. Energy for forming phosphodiester bonds comes from hydrolyzing the bond between the first and second phosphate groups of each dNTP, releasing pyrophosphate (P-P).

Temperature cycling in PCR
The graph below shows the temperature changes during PCR cycles:
Taq polymerase: a thermostable enzyme
Taq polymerase is derived from the bacterium Thermus aquaticus, discovered in hot springs at Yellowstone Park in . This enzyme remains active at the high temperatures used during PCR, unlike DNA polymerases from most organisms which would denature above .
The thermostability of Taq polymerase is crucial because it means the enzyme doesn't need to be replaced after each denaturation step - it can function throughout multiple temperature cycles. This makes PCR an automated and efficient process.
Amplification through repeated cycles
After each cycle, the DNA is heated again to separate strands, primers bind, and Taq polymerase creates new complementary strands. A single DNA molecule can produce billions of copies through repeated cycling.
Worked Example: Calculating DNA Amplification
The number of DNA molecules increases exponentially: after cycles, theoretically copies exist.
After cycles:
After cycles:
After cycles:
This exponential growth explains how PCR can rapidly amplify even tiny amounts of DNA.
Multiplex PCR and inhibitors
Multiplex PCR involves simultaneously amplifying multiple DNA sequences in a single reaction by using more than one pair of primers.
PCR Inhibitors to Be Aware Of:
Certain substances can inhibit PCR reactions:
- Ionic detergents and gel loading dyes from DNA extraction
- Proteinase K (an enzyme used in DNA extraction that can break down Taq polymerase if not removed)
- Components in blood such as haemoglobin and the anti-clotting agent heparin
Proper DNA purification is essential to remove these inhibitors before PCR.
PCR vs natural DNA replication
Key Differences Between PCR and Natural DNA Replication:
- PCR operates at much higher temperatures ( vs below )
- PCR uses DNA primers rather than RNA primers
- PCR amplifies only specific short DNA sequences, not entire chromosomes
- PCR requires thermostable DNA polymerase
- PCR occurs in small plastic tubes in a thermal cycler, not within living cells
Gel electrophoresis
Gel electrophoresis is the separation of charged molecules by differential movement through a gel in an electric field. The degree of movement depends on the mass of the molecules and their net charge.
This technique is used to separate and identify proteins and DNA fragments, similar to how chromatography separates chemical mixtures.
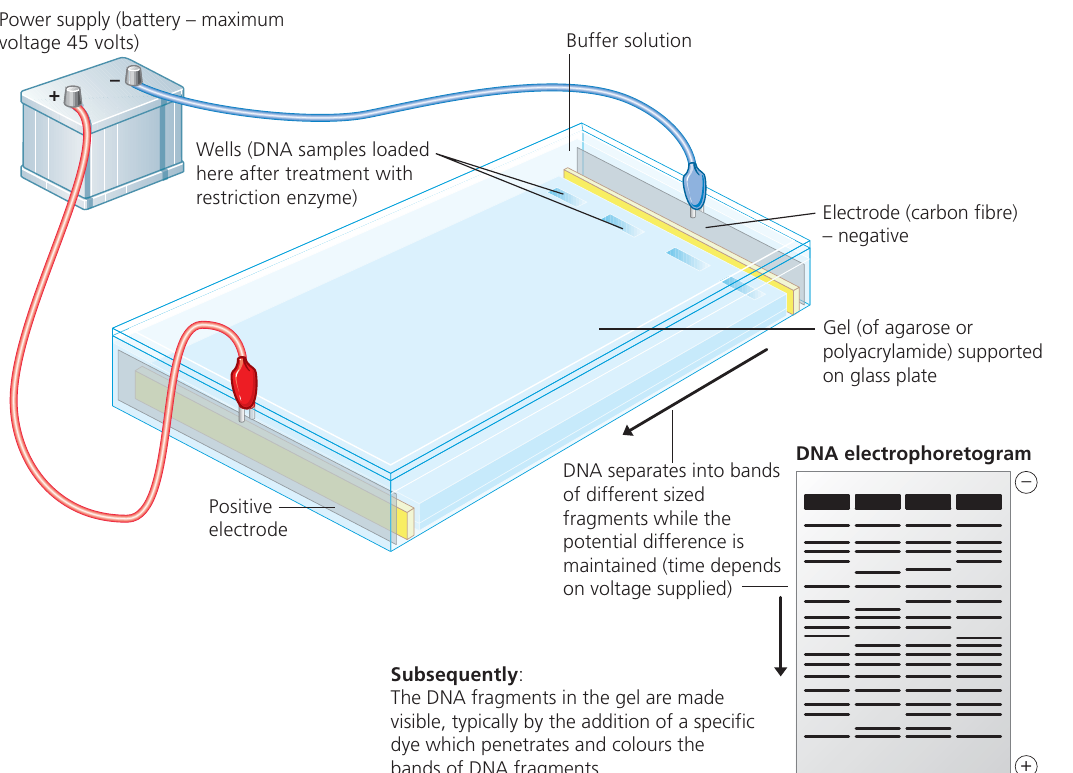
Principles of electrophoresis
The basic setup involves:
- A gel (acting as a molecular sieve) supported on a glass or plastic plate
- Wells cut into the gel where samples are loaded
- Buffer solution covering the gel to conduct electricity
- Electrodes connected to a power supply creating an electric field
The anode is the positive electrode that attracts negatively charged molecules, while the cathode is the negative electrode that attracts positively charged molecules.
Electrophoresis of proteins
Protein charge depends on the ionization of R groups on amino acid residues:
- Some R groups are positively charged (–NH₃⁺)
- Some are negatively charged (–COO⁻)
- Some are uncharged
Whether these groups are charged depends on pH, so electrophoresis is performed in buffer solution to maintain constant pH.
Polyacrylamide gel electrophoresis (PAGE) is commonly used for protein separation. Proteins are typically treated with:
- A reducing agent (mercaptoethanol) to break disulfide bonds
- Sodium dodecyl sulfate (SDS) which denatures proteins into negatively charged rod shapes
This SDS-PAGE treatment ensures proteins separate primarily by size rather than charge - smaller proteins move faster through the gel toward the anode.
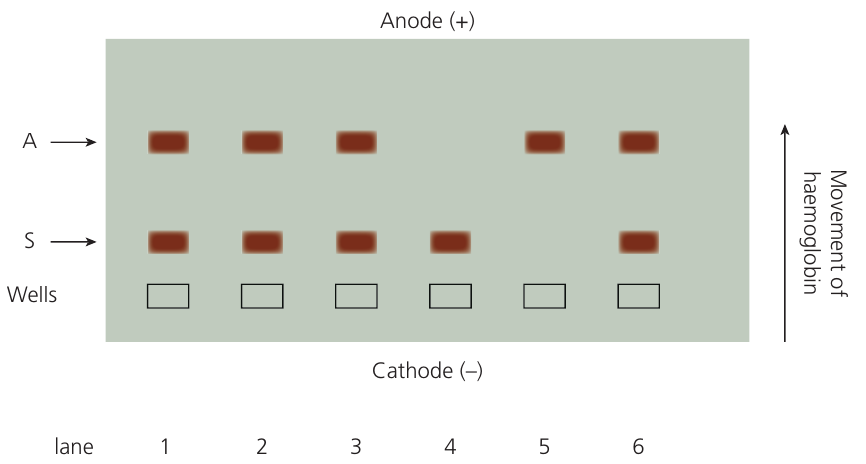
Application: detecting sickle cell haemoglobin
Gel electrophoresis can distinguish between normal and sickle cell variants of β globin.
Worked Example: Detecting Sickle Cell Anaemia
In sickle cell anaemia, the amino acid at position changes from valine (with an uncharged R group) to glutamic acid (with a negatively charged R group).
Effect on electrophoresis:
This substitution gives sickle cell haemoglobin a slightly lower negative charge than normal haemoglobin, causing it to move a shorter distance through the gel.
Results interpretation:
The technique can identify:
- Individuals with sickle cell anaemia (only sickle cell haemoglobin)
- Carriers with sickle cell trait (both normal and sickle cell haemoglobin)
- Unaffected individuals (only normal haemoglobin)
Electrophoresis of DNA
All DNA fragments carry negative charges due to phosphate groups in the sugar-phosphate backbone. In DNA electrophoresis, fragments move through the gel toward the anode.
The distance travelled by a DNA fragment is inversely proportional to its length: shorter fragments move faster and travel further than longer fragments. Fragments of identical size migrate the same distance and form distinct bands.

Procedure for DNA gel electrophoresis
The flowchart below outlines the key steps:
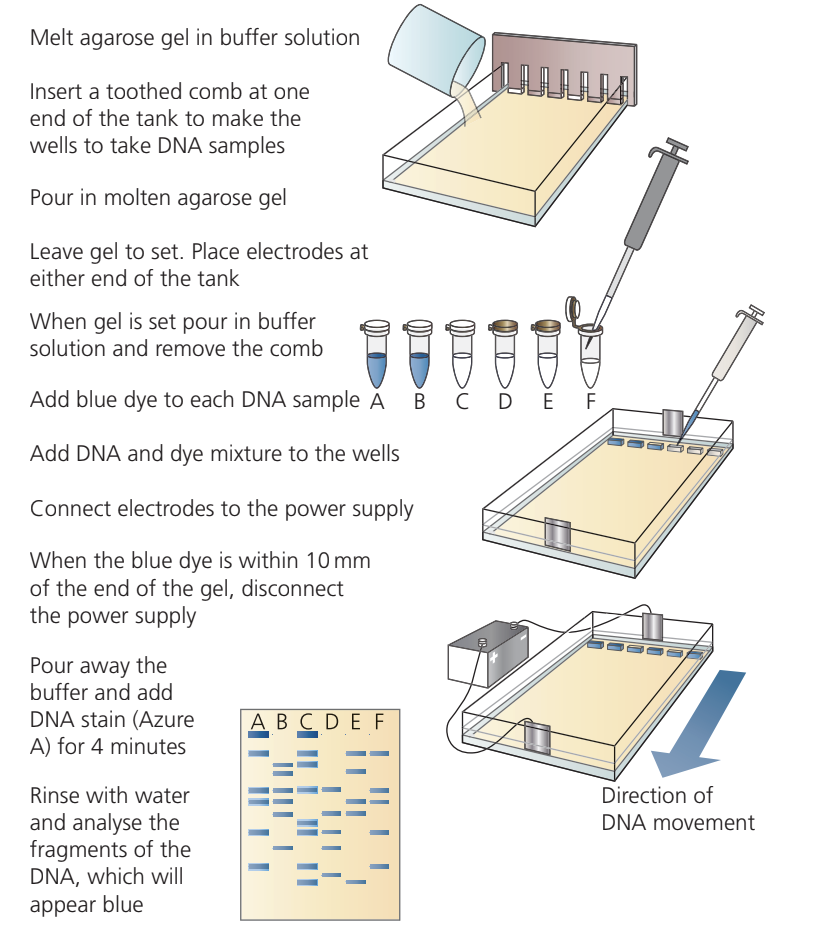
Key Steps in DNA Gel Electrophoresis:
- Prepare agarose gel by melting in buffer solution
- Pour gel into casting tray with comb to create wells
- Allow gel to set, then remove comb and cover with buffer
- Add tracking dye to DNA samples
- Load DNA-dye mixture into wells using micropipettes
- Connect electrodes and apply voltage (maximum V)
- Run until tracking dye approaches the end of the gel
- Stop electrophoresis and stain DNA (commonly with Azure A or ethidium bromide)
- Visualize and analyze the banding pattern
The tracking dye moves slightly ahead of the smallest DNA fragments, allowing you to monitor progress and stop before DNA runs off the gel.
Capillary flow electrophoresis
This automated technique combines DNA separation with fluorescent detection. DNA fragments tagged with fluorescent markers are separated in narrow capillary tubes. As each fragment passes a laser, it fluoresces, and a detector records the signal.
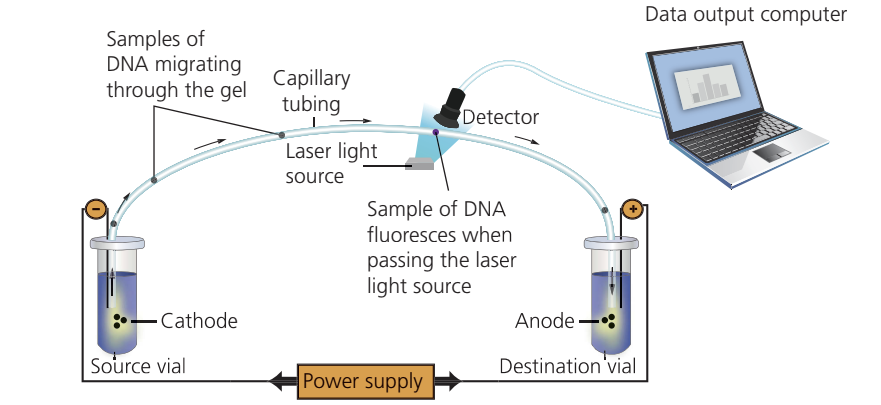
This method dramatically increased sequencing speed and was used in first-generation DNA sequencing machines, making large-scale genome projects feasible.
DNA sequencing
DNA sequencing determines the order of nucleotide bases (A, T, C, G) in DNA samples by detecting bases as they are added to a template strand by polymerase enzyme.
Chain-termination method
Developed in the s, this method involves replicating short single-stranded DNA fragments similar to PCR, but with a key difference: the reaction mixture contains modified nucleotides called dideoxynucleotides.
These modified nucleotides terminate DNA synthesis at specific bases (A, T, C, or G), producing multiple DNA copies of varying lengths. These fragments can be separated by electrophoresis based on their different masses.
High-throughput sequencing
High-throughput sequencing refers to any method that determines base sequences in DNA samples (including whole genomes) rapidly.
The development of sequencing machines with sets of capillary flow electrophoresis apparatus dramatically increased sequencing speed, making whole-genome sequencing feasible. This technological advance was crucial for completing projects like the Human Genome Project.
Approaches to genome sequencing
Traditional mapping methods used genetic crosses and pedigree analysis to map gene locations on chromosomes, particularly useful for studying genetic diseases like Huntington's disease and haemophilia.
Shotgun sequencing involves:
- Sequencing random DNA fragments without knowing their genomic location
- Comparing sequences to find overlapping regions
- Assembling fragments like pieces of a linear jigsaw puzzle
Worked Example: First Application of Shotgun Sequencing
This approach was first used to sequence the genome of Haemophilus influenzae (a bacterium with a circular chromosome of nearly million base pairs).
Achievement:
By , shotgun sequencing combined with traditional methods produced a draft sequence of the human genome, containing approximately billion base pairs.
Key Points to Remember:
-
PCR amplifies specific DNA sequences through three repeated stages: denaturation at , annealing at , and extension at
-
Primers are essential in PCR - they identify where synthesis begins and provide the double-stranded region needed by DNA polymerase
-
Taq polymerase is thermostable - it remains active at high temperatures, allowing it to function throughout multiple PCR cycles without replacement
-
Gel electrophoresis separates molecules by size and charge - proteins separate based on net charge (in PAGE), while DNA fragments separate based on length (shorter = further migration)
-
The anode attracts negative charges - DNA always moves toward the anode because phosphate groups make it negatively charged; proteins can move toward either electrode depending on their charge
-
DNA amplification is exponential - after PCR cycles, you have copies of the target sequence
-
SDS-PAGE treatment ensures proteins separate primarily by size rather than charge
-
High-throughput sequencing made whole-genome projects feasible through automation and increased processing speed