DNA Sequencing and Profiling (OCR A-Level Biology A): Revision Notes
DNA Sequencing and Profiling
DNA sequencing techniques
DNA sequencing determines the specific order of nucleotide bases (adenine, thymine, cytosine, and guanine) along a DNA molecule. The sequence information reveals the genetic instructions encoded within genes and provides insights into evolutionary relationships, genetic variation, and disease mechanisms.
Chain-termination method
The chain-termination method, also known as Sanger sequencing, was developed in the 1970s and represented a breakthrough in molecular biology. This technique works by copying short fragments of single-stranded DNA template, similar to the polymerase chain reaction (PCR).
The key innovation involves adding modified nucleotides called dideoxynucleotides to the reaction mixture. These modified nucleotides lack the 3' hydroxyl group needed to form the next phosphodiester bond, so when incorporated, they terminate DNA synthesis at that position. By including different dideoxynucleotides for each of the four bases (A, T, C, G), the reaction produces multiple DNA copies of varying lengths, each ending at a specific base position.
These partially synthesized DNA molecules can then be separated by electrophoresis based on their different masses. Initially, this was a time-consuming process, but speed increased dramatically with the development of high-throughput sequencing machines containing sets of capillary flow electrophoresis apparatus running in parallel. This technological advance made it practical to sequence entire genomes.
Approaches to genome sequencing
Two main strategies emerged for sequencing complete genomes:
Traditional genetic mapping began by identifying the chromosomal locations of genes using data from genetic crosses and pedigree analysis of families affected by genetic diseases such as Huntington's disease and haemophilia. This approach provided a framework for understanding genome organization.
Shotgun sequencing offered a complementary approach by sequencing random DNA fragments without prior knowledge of their genomic location. After sequencing many fragments, bioinformatics tools compared the sequences to identify overlapping regions, allowing the fragments to be assembled like pieces in a very long, linear jigsaw puzzle. This method was first successfully applied to sequence the nearly million base pair circular chromosome of the bacterium Haemophilus influenzae. By 2001, combining shotgun sequencing with traditional mapping approaches produced the first draft of the human genome sequence.
Next-generation sequencing
The chain-termination method has now been superseded by next-generation sequencing (NGS) technologies. These advanced methods can sequence thousands to millions of DNA molecules simultaneously without requiring separate electrophoresis steps. Different detection systems identify the bases as they are incorporated during DNA synthesis.
One example is pyrosequencing, which uses the enzyme luciferase to emit flashes of light when pyrophosphate (P-P) is released during nucleotide addition to the growing DNA strand. Each flash indicates that a specific base has been incorporated, allowing the sequence to be read in real time.

Advantages of Next-Generation Sequencing:
Next-generation methods operate to times faster than previous techniques, reducing the cost of sequencing million nucleotides ( megabase pairs) to as little as 0.1% of the cost of chain termination. Complete genomes can now be sequenced for less than US$1000, making genomic analysis accessible for research and clinical applications.
Timeline of DNA sequencing developments
| Year | Development |
|---|---|
| 1975 | Chain-termination method developed by Fred Sanger and Andrew Coulson in the UK; chemical degradation method by Alan Maxam and Walter Gilbert in the USA |
| 1977 | First complete genome sequenced: bacteriophage φX174 with nucleotides |
| 1990s | Development of capillary flow electrophoresis with appropriate DNA detection systems; shotgun approach used for Haemophilus influenzae genome |
| 1996 | Yeast (Saccharomyces cerevisiae) genome sequenced – first eukaryote completed |
| 1998 | Sequencing machines with capillary sequencers developed, termed high-throughput sequencing |
| 2001 | First draft of human genome published (complete version in 2004) |
| 2005 | First next-generation sequencing machine became commercially available |
| 2012 | Tomato (Solanum lycopersicum) genome published using both chain-termination and next-generation methods – probably the last genome sequenced using Sanger sequencing |
| 2015 | Genome of extinct woolly mammoth sequenced |
Nanopore sequencing
An emerging technology promises to revolutionize DNA sequencing by making it portable and even more accessible. Nanopore sequencing works by threading a DNA molecule through a tiny protein pore embedded in a lipid bilayer membrane. As the DNA passes through the pore, ions flow through the channel, creating an electrical current. Each of the four bases (A, T, C, G) alters the current flow in a distinctive way, allowing the sequence to be determined in real time.
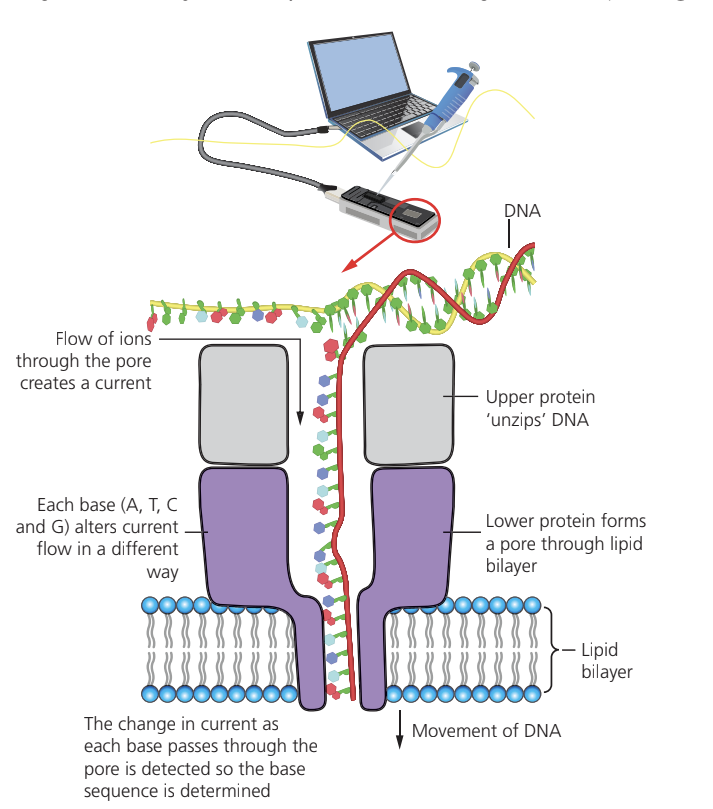
This method can potentially be carried out rapidly almost anywhere, not requiring expensive laboratory facilities. The technology is still being refined and tested, but when fully developed, it should make sequence data available cheaply and easily for diverse applications across biology.
Applications of whole-genome sequencing
The reduced costs of next-generation sequencing have enabled genomes from numerous species to be sequenced. Comparing whole genomes allows scientists to locate the same or similar genes across different species and analyze evolutionary relationships.
Sequencing genomes from many individuals within the same species reveals information about genetic variation within populations. This has been applied to humans and other mammals, particularly domesticated animals (horse, cattle, pig, yak) and endangered species such as giant pandas. Important crop plants including rice, papaya, cabbage, potato, wheat, and tomato have also had their genomes sequenced.
Synthetic biology
Understanding genome sequences enables researchers to manipulate genetic systems in novel ways. Synthetic biology is a new science that involves designing new biological systems and redesigning existing systems using knowledge of nucleic acids and cell biology.
Scientists have used genome information to perform "knockout" experiments, systematically removing genes from simple organisms like Mycoplasma to determine the minimum number required for life. Similar studies in other prokaryotes and in yeast have revealed essential gene sets.
Researchers have also assembled synthetic genomes from scratch, creating artificial organisms. Gene sequencing allows scientists to design completely new base sequences and insert them into bacterial or eukaryotic cells using genetic engineering techniques. Because they understand the genetic code, they can predict the amino acid sequence encoded by any DNA sequence and even predict how the resulting proteins will fold into specific three-dimensional structures.
This capability allows for the production of novel molecules, such as enzymes for medicinal drug production. Existing genes can be modified to improve protein function, as demonstrated by the various forms of human insulin now available for diabetes treatment. Entire cellular systems can be redesigned to make genetically modified organisms work more efficiently in producing desired proteins.
Epidemiology applications
Whole-genome sequencing of pathogens has transformed epidemiology – the study of disease spread in populations. Sequencing enables epidemiologists to identify specific strains infecting people, livestock, and crops, determining the most appropriate control methods. For example, genome sequencing of the Ebola virus contributed significantly to understanding disease transmission during the 2015 West African epidemic, helping to track the spread and implement targeted interventions.
DNA profiling
Genome sequencing has revealed extensive variation in DNA between individuals within a species. This variation makes it possible to identify individuals with very high accuracy, since the probability of two individuals having identical DNA is extremely small (unless they are clones).
Repeated sequences in DNA
A distinctive feature of genomes is the presence of nucleotide sequences that are repeated, often many times. These repeated regions can be treated as genetic markers, with the variable number of repeats functioning like different alleles. Because these regions show high variability between individuals, they represent examples of genetic polymorphism.
Before high-throughput sequencing became available, individual identification relied on detecting differences in phenotypes, such as using protein electrophoresis to identify sickle cell anaemia carriers or using antibodies to determine blood groups. The discovery of repeated DNA sequences led to methods of genetic profiling (often called genetic fingerprinting).
Minisatellites were among the first markers used. These sequences, – base pairs in length, repeat at various chromosomal locations and are inherited from both parents. They can be cut by restriction enzymes and the fragment lengths determined by gel electrophoresis.
Short tandem repeats
The use of minisatellites has been largely replaced by short tandem repeats (STRs), also known as microsatellites. These are much shorter than minisatellites, consisting of – nucleotides repeated – times. For instance, the sequence CACACA might be repeated between five and twenty times at a particular genomic location.
The number of repeats at any STR locus varies between individuals. This variability arises from the inaccuracy of DNA polymerase when copying these repetitive regions during replication. Although the error rate is generally very low, occasional mistakes lead to changes in the number of repeat units, creating new alleles.
STR markers can be classified as:
- Simple: identical-length repeats
- Compound: two or more adjacent repeats
- Complex: several different length repeats
STRs occur on all autosomal chromosomes as well as both X and Y sex chromosomes. The CAG repeat sequence in the huntingtin gene represents one example of an STR. In DNA profiling, the alleles at selected STR loci are determined. Many of the most useful STRs for identification purposes are located in non-coding DNA between structural genes.
Forensic DNA profiling
Forensic scientists use DNA profiling to identify crime victims, suspects in criminal cases, disaster victims, and missing people. The European DNA 17 profile consists of STR loci plus markers that identify sex, providing a highly discriminating identification system.
Characteristics of forensic STR markers
STRs selected for forensic applications typically have four- or five-nucleotide repeats. These were chosen because they:
- Show high degrees of polymorphism, providing excellent discrimination between individuals
- Are robust and suffer only low levels of environmental degradation
- Provide high-quality, error-free data
- Represent discrete alleles that are clearly distinguishable from one another
- Require only small sample amounts (as little as picograms from about cells)
- Are located in non-coding DNA, so no selective pressure acts against any alleles
- Can be efficiently amplified by PCR, including multiplex PCR of multiple loci simultaneously
- Produce low levels of artefact formation during amplification
STR analysis process
PCR amplifies the DNA samples to produce many copies of the regions containing the STRs. The STRs are labelled with fluorescent dyes, and the sequences are separated by gel electrophoresis or detected during PCR using four different fluorescent colours (typically red, green, yellow, and blue). A laser scanner detects the fluorescence as each fragment passes the detector.
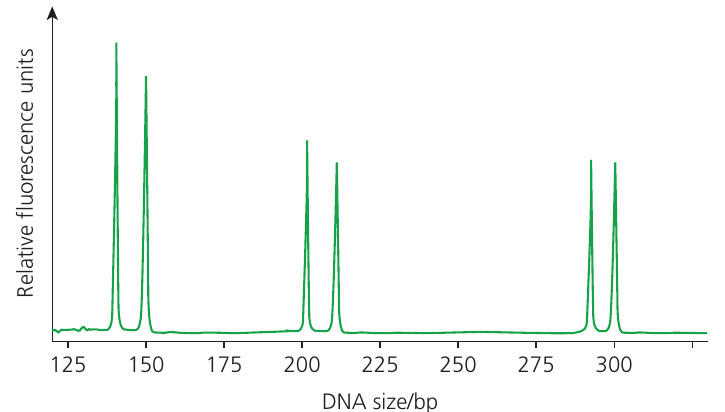
STR profiles display results in multiple colour channels using a four-colour fluorescent system, with different channels showing different sets of STR loci. A typical printout shows the individual's sex and two peaks for most STR loci. The two peaks represent the two alleles inherited from each parent. Most people are heterozygous, having different numbers of repeats in the alleles inherited from their mother and father.
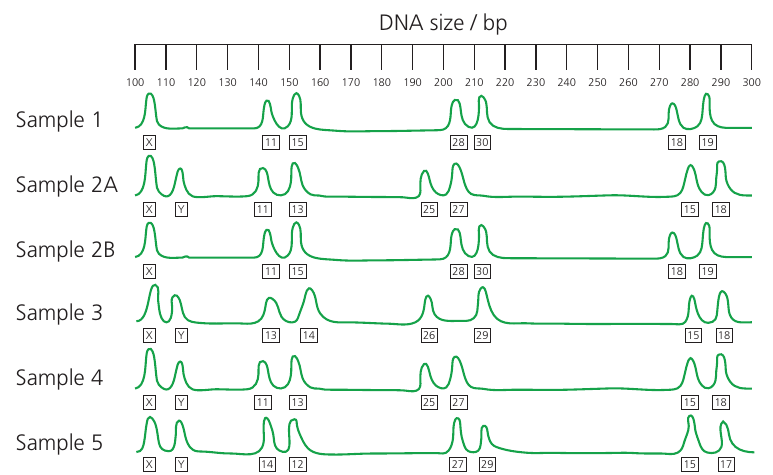
Forensic Application: Sexual Assault Case
In a sexual assault case, samples might include vaginal swabs from the victim and buccal (cheek) swabs from the victim and potential suspects. By comparing the STR profiles, forensic scientists can determine which DNA belongs to the victim and which to the perpetrator, and identify matches with suspect samples.
DNA profiling and disease risk
Genetic testing
DNA testing or genetic testing detects the risk of genetic diseases or disorders. DNA samples typically come from blood samples or buccal swabs (cotton swabs wiped inside the cheek to collect epithelial cells). Almost all molecular genetic tests use PCR to amplify specific DNA regions.
Because PCR is highly selective, it can amplify just the DNA section needed for testing. However, PCR typically amplifies only short sequences (up to about bases). Since most genes are larger than this, multiplex PCR reactions are needed to copy entire genes.
Genetic tests exist for many monogenic diseases, including:
- Cystic fibrosis
- Huntington's disease
- Sickle cell anaemia
- β-thalassaemia
- Haemophilia A and B
- Severe combined immunodeficiency syndrome
- Some forms of breast cancer
These diseases result from inherited mutations that run in families. When someone tests positive for a disease-causing mutation, genetic tests are often offered to other family members. Some people know a particular disease runs in their family and choose genetic testing. However, in some cases (such as haemophilia), up to 40% of affected individuals come from families with no previous disease history.
Understanding Genetic Risk
Inheriting a disease-associated allele does not always mean the disease will develop. For example, people who inherit the dominant allele for Huntington's disease do not always develop symptoms. Similarly, people who inherit the BRCA1 or BRCA2 genes (autosomal dominant) have an increased risk of developing breast cancer but do not necessarily develop the disease.
Single nucleotide polymorphisms
Single nucleotide substitutions cause most genetic diseases. Single nucleotide polymorphisms (SNPs), pronounced "snips", are the simplest and most common type of genetic variation, comprising around 90% of genetic variation in humans. They occur during meiosis when DNA is replicated.
A SNP represents a change of a single base at a specific position in the DNA sequence. For example, a cytosine might be replaced with a thymine in a particular DNA stretch. This does not change the DNA length, so if the SNP falls in an exon, there is no effect on the gene's reading frame during protein synthesis.
About million SNPs exist in the human genome, with one found approximately every – base pairs. They serve as useful biological markers of disease. Each SNP can consist of up to four alleles, because there are four possible bases (A, T, C, G) at each nucleotide position. SNP analysis requires DNA sequencing techniques to establish which specific base is present at each SNP location.
Different human genome sequencing projects have revealed that individuals have between and million SNPs compared to the reference sequence. So far, over million SNPs have been discovered from human genome sequencing projects.
Personalised medicine
The availability of inexpensive next-generation sequencing techniques, such as nanopore sequencing, will allow doctors to determine the sequence of specific genome regions. This information enables the prescription of drugs that will be effective for people with particular genotypes. This approach represents personalised medicine, where treatment is tailored to each individual's genetic makeup, potentially improving outcomes and reducing adverse drug reactions.
Key Points to Remember:
- DNA sequencing determines the order of nucleotide bases along DNA molecules, with methods evolving from chain-termination to next-generation techniques
- Capillary flow electrophoresis automates DNA separation and detection using fluorescent markers and laser detection
- Next-generation sequencing can sequence thousands to millions of DNA molecules simultaneously at a fraction of the cost of earlier methods
- STRs (short tandem repeats) are – nucleotide sequences repeated – times, showing high variation between individuals
- Forensic DNA profiling uses STR loci plus sex markers to identify individuals with high accuracy
- SNPs (single nucleotide polymorphisms) are single-base changes representing the most common genetic variation, with approximately million present in the human genome
- Whole-genome sequencing enables comparative genomics, synthetic biology, epidemiology, and personalised medicine applications