Recombination and Transformation (VCE SSCE Biology): Revision Notes
Recombination and Transformation
Introduction
Diabetes affects approximately 1.2 million Australians. This condition occurs when the pancreas cannot adequately produce insulin, a hormone responsible for glucose uptake from the bloodstream into cells. Whilst there is currently no cure for diabetes, the condition can be managed through regular insulin injections. This raises an important question: how can we produce enough insulin for 5% of the Australian population to use daily?
The answer lies in genetic engineering. Through bacterial transformation, scientists have developed methods to make bacteria produce human insulin and other vital proteins.
Why transform bacteria?
Bacteria are simple prokaryotic organisms that contain two types of genetic material. They have a main circular chromosome and smaller, independent loops of DNA called plasmids.
Plasmid – a small, circular loop of DNA separate from the chromosome, typically found in bacteria.
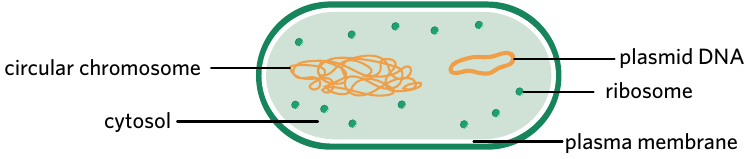
Plasmids replicate independently from the bacterial chromosome. The number of plasmids varies between individual bacteria - some may contain many plasmids whilst others have none. These plasmids can range in length up to 200,000 base pairs (200 kbp).
The independent replication of plasmids makes them extremely useful for genetic modification. Unlike chromosomal DNA, plasmids can be easily removed, modified, and reintroduced into bacteria without disrupting essential cellular functions.
Scientists can edit plasmids to include target genes of interest. Once a plasmid has been modified to incorporate a foreign gene, it becomes a recombinant plasmid. When bacteria take up these recombinant plasmids from their environment through a process called bacterial transformation, they gain the ability to produce specific proteins encoded by the foreign gene.
Recombinant plasmid – a circular DNA vector that is ligated to incorporate a gene of interest.
Bacterial transformation – the process by which bacteria take up foreign DNA from their environment. Scientists use this process to introduce recombinant plasmids into bacteria.
Genetic modification – the manipulation of an organism's genetic material using biotechnology.
This technology has revolutionized modern medicine and agriculture by enabling cheaper and more efficient production methods. Bacterial transformation is now used to produce many important proteins, including:
- Insulin to manage diabetes
- Erythropoietin to treat anaemia
- Chymosin for cheese production (previously extracted from calf stomachs)
- Interferon to treat some cancers
- Growth hormone to manage growth disorders
- Hepatitis B surface antigen for vaccines
- Alpha-amylase for ethanol and high fructose corn syrup production
Making a recombinant plasmid
Creating recombinant plasmids requires genetic engineers to insert foreign DNA into bacterial plasmids. Once bacteria take up these modified plasmids, they will express the protein encoded by the foreign DNA. This process requires four key components:
- Gene of interest
- Plasmid vector
- Restriction endonuclease
- DNA ligase
Gene of interest
The gene of interest (also called the desired gene) is a DNA sequence encoding the protein scientists wish to produce.
Gene of interest – a gene scientists want to be expressed in recombinant bacteria. This gene often encodes a protein we wish to produce in commercial quantities.
The gene is typically isolated from human DNA and amplified using the polymerase chain reaction (PCR). An important feature is that bacteria can use human DNA to produce identical human proteins because the genetic code is universal. For example, a CUU codon codes for the amino acid leucine in all organisms, whether human or bacterial.
Critical requirement: Removal of introns
The gene of interest must not contain introns before insertion into the plasmid. This is because prokaryotic gene expression does not involve RNA processing like eukaryotic systems. In simple terms, bacteria lack the cellular machinery to remove introns from mRNA, so they cannot process genes containing introns.
Introns are typically removed using two methods:
- Synthetic DNA – Genes are manufactured artificially in a laboratory using a DNA synthesizer. Introns are simply not included when the gene is created.
- Copy DNA (cDNA) – An enzyme called reverse transcriptase converts mature mRNA (which already has introns removed) back into DNA. Since the mRNA template lacks introns, the resulting cDNA also lacks introns.
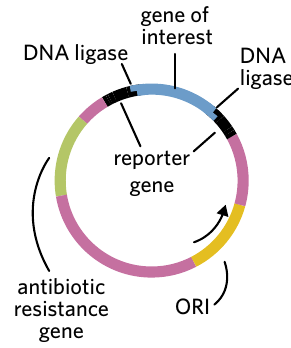
Plasmid vector
A plasmid vector is a carefully designed piece of circular DNA that has been modified to be ideal for bacterial transformation experiments.
Plasmid vector – a piece of circular DNA that is modified to be an ideal vector for bacterial transformation experiments.
Vector – a means of introducing foreign DNA into an organism. Plasmids are a popular vector in bacterial transformation.
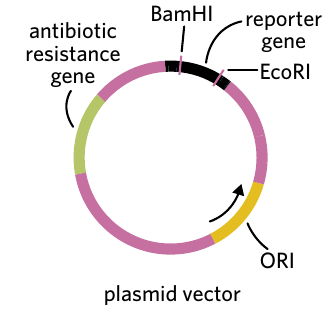
Most plasmid vectors contain four essential DNA sequences:
1. Restriction endonuclease sites
These are specific DNA sequences that can be recognized and cut by restriction enzymes. They provide locations where the gene of interest can be inserted into the plasmid. Common examples include BamHI and EcoRI restriction sites.
2. Antibiotic resistance genes
These genes allow bacteria carrying the plasmid to survive in the presence of specific antibiotics. For example:
- ampR confers resistance to ampicillin
- tetR confers resistance to tetracycline
Antibiotic resistance gene – gene which confers antibiotic resistance.
3. Origin of replication (ORI)
This is a DNA sequence that signals where DNA replication should begin in bacterial cells. It ensures the plasmid can be copied when bacteria divide.
Origin of replication (ORI) – a sequence found in prokaryotes that signals the start site of DNA replication.
4. Reporter gene
These genes produce easily observable characteristics that help identify successful genetic modification. Reporter genes have recognizable phenotypes that can be used to distinguish recombinant plasmids from non-recombinant plasmids.
Reporter gene – gene with an easily identifiable phenotype that can be used to identify whether a plasmid has taken up the gene of interest.
An example is the gfp gene, which produces green fluorescent protein that glows green under UV light.
Important design requirement: For a plasmid vector to be effective, it must contain two genes encoding observable traits (such as two antibiotic resistance genes, or one antibiotic resistance gene plus one reporter gene). Additionally, one of these genes must contain the restriction site where the gene of interest will be inserted. This design allows scientists to distinguish between successful and unsuccessful genetic modifications in later steps.
Restriction endonuclease
Restriction endonuclease (also called restriction enzyme) – any enzyme that acts like molecular scissors to cut nucleic acid strands at specific recognition sites.
Both the gene of interest and the plasmid vector are cut using the same restriction endonuclease. This creates identical "sticky ends" on the gene and the plasmid, allowing them to join together.
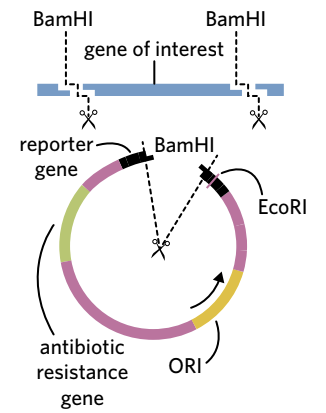
For example, the restriction enzyme BamHI can be used to:
- Cut out the gene of interest from source DNA
- Create an opening in the plasmid vector (specifically within the reporter gene)
The overhanging nucleotides created by the enzyme on the gene of interest are complementary to the overhanging nucleotides on the plasmid. This complementarity allows them to form hydrogen bonds easily. Whilst blunt-end restriction enzymes can also be used, they are less targeted because any blunt end can bond with any other blunt end.
DNA ligase
Ligase – an enzyme that joins molecules, including DNA or RNA, together by catalysing the formation of phosphodiester bonds.
DNA ligase is the final component needed to create a recombinant plasmid. This enzyme joins the gene of interest to the plasmid vector by forming strong phosphodiester bonds in the sugar-phosphate backbone of DNA.
The result is a circular piece of DNA called a recombinant plasmid. However, the process is not perfectly efficient.
Most plasmids will simply join back to themselves without taking up the gene of interest - these are called non-recombinant plasmids. The final mixture therefore contains both recombinant and non-recombinant plasmids. Distinguishing between these two types relies on the reporter gene, but this can only be done after bacteria have taken up the plasmids through transformation.
Transforming bacteria
Uptake of recombinant plasmids
Many bacteria can naturally take up free-floating DNA from their environment and incorporate it into their cytoplasm through transformation. Scientists exploit this natural ability to introduce recombinant plasmids into bacterial cells.
Two main methods are used to enhance plasmid uptake:
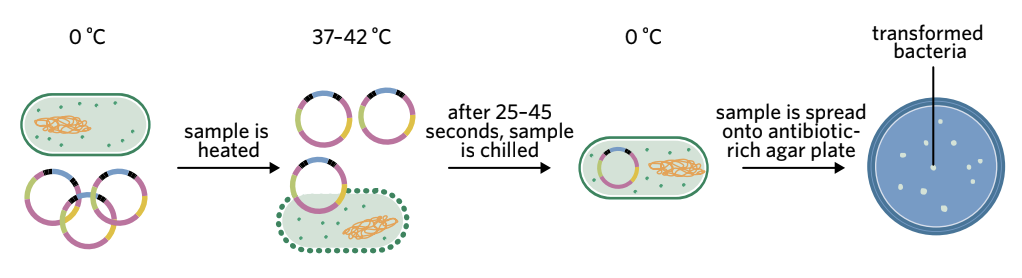
Heat shock method
Heat shock – a method that involves rapidly increasing and decreasing the temperature to increase membrane permeability in order to enhance the likelihood of bacterial transformation.
The heat shock process follows these steps:
- Bacteria and plasmids are placed in a calcium ion solution on ice (0°C)
- The positive calcium ions make the negatively charged bacterial membrane more permeable to negatively charged plasmid DNA
- The solution is rapidly heated to 37-42°C for 25-45 seconds
- The solution is immediately returned to ice (0°C)
- The sudden temperature change increases membrane permeability, allowing plasmids to cross the membrane
- The mixture is spread onto antibiotic-rich agar plates
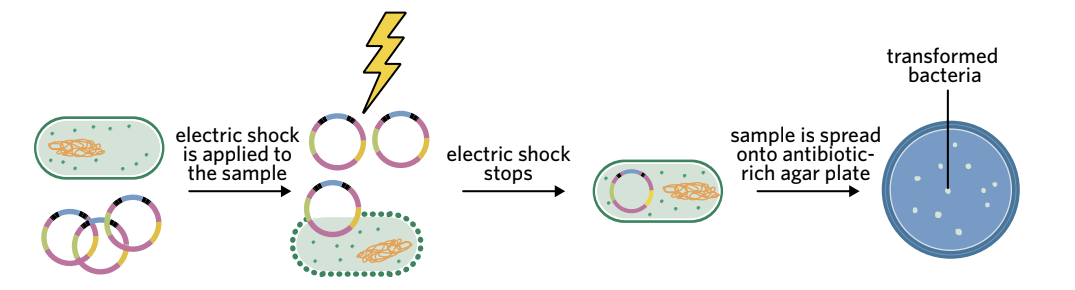
Electroporation method
Electroporation – a method that involves delivering an electric shock to bacterial membranes to increase their membrane permeability and increase the likelihood of bacterial transformation.
This method is similar to heat shock but uses electricity instead of temperature:
- Bacteria and plasmids are mixed in solution
- An electrical current is passed through the solution
- The electric shock makes the membrane more permeable
- Plasmids can cross the phospholipid bilayer
- The electric shock is stopped
- The mixture is spread onto antibiotic-rich agar plates
Antibiotic selection
After transformation, the bacterial mixture contains both transformed bacteria (which took up plasmids) and untransformed bacteria (which did not). These need to be separated.
The mixture is cultured onto agar plates containing a specific antibiotic. Only transformed bacteria possess the antibiotic resistance gene from the plasmid, so only they can survive on the antibiotic-rich plate. All untransformed bacteria are killed by the antibiotic.
Each visible colony on the plate represents a successful transformation event. A single bacterium took up a plasmid, survived the antibiotic exposure, multiplied, and formed a colony of genetically identical daughter cells.
Distinguishing recombinant from non-recombinant plasmids
Transformed bacteria can contain either recombinant plasmids (with the gene of interest) or non-recombinant plasmids (which joined back to themselves). These must be distinguished from each other using the reporter gene.
In recombinant plasmids, the reporter gene was cut by the restriction enzyme and became the insertion site for the gene of interest. This means the reporter gene is disrupted and non-functional.
Example: Using the gfp reporter gene
The gfp gene encodes green fluorescent protein, which glows green under UV light.
- Non-recombinant plasmids: The gfp gene is intact and continuous. Bacteria with these plasmids produce functional green fluorescent protein and glow green under UV light.
- Recombinant plasmids: The gfp gene is interrupted by the inserted gene of interest. The gene is non-continuous and cannot produce functional fluorescent protein. Bacteria with these plasmids do NOT glow under UV light.
Scientists can therefore identify bacteria containing recombinant plasmids by selecting colonies that do not fluoresce under UV light.
Protein production and extraction
Once bacteria containing recombinant plasmids are identified, they are:
- Cultured to grow into large populations
- Induced to express the gene of interest and produce the target protein
- Harvested and processed to extract the desired protein
- Purified to remove other bacterial proteins
The purified protein can then be used in medicine, research, or industry.
Insulin production
Insulin – a hormone secreted by the pancreas to control blood glucose levels.
Diabetes – a disease where the body cannot properly produce or respond to insulin.
Background
Before genetic engineering techniques were developed, diabetic patients received insulin extracted from the pancreases of pigs (porcine insulin) or cows (bovine insulin). These animal insulins have similar structures to human insulin, but this method had significant drawbacks:
- Required killing many animals to extract insulin from their pancreases
- Less effective at regulating blood glucose than human insulin
- Could cause immune reactions in some patients
- Expensive and inefficient
In the late 1970s, scientists successfully produced human insulin in bacteria using recombinant plasmids. This method has become the standard approach because it is significantly cheaper, more effective, and does not require animal sacrifice.
Insulin structure
Insulin has a quaternary protein structure consisting of two polypeptide chains:
- Alpha subunit (A chain)
- Beta subunit (B chain)
These two chains are joined together by disulphide bridges to form functional insulin.
Critical point: Because insulin consists of two separate chains, producing it requires TWO different recombinant plasmids and TWO different transformed bacterial cultures - one producing the alpha subunit and one producing the beta subunit. The chains must first be produced separately, then combined and folded together to create functional insulin.
The insulin production process
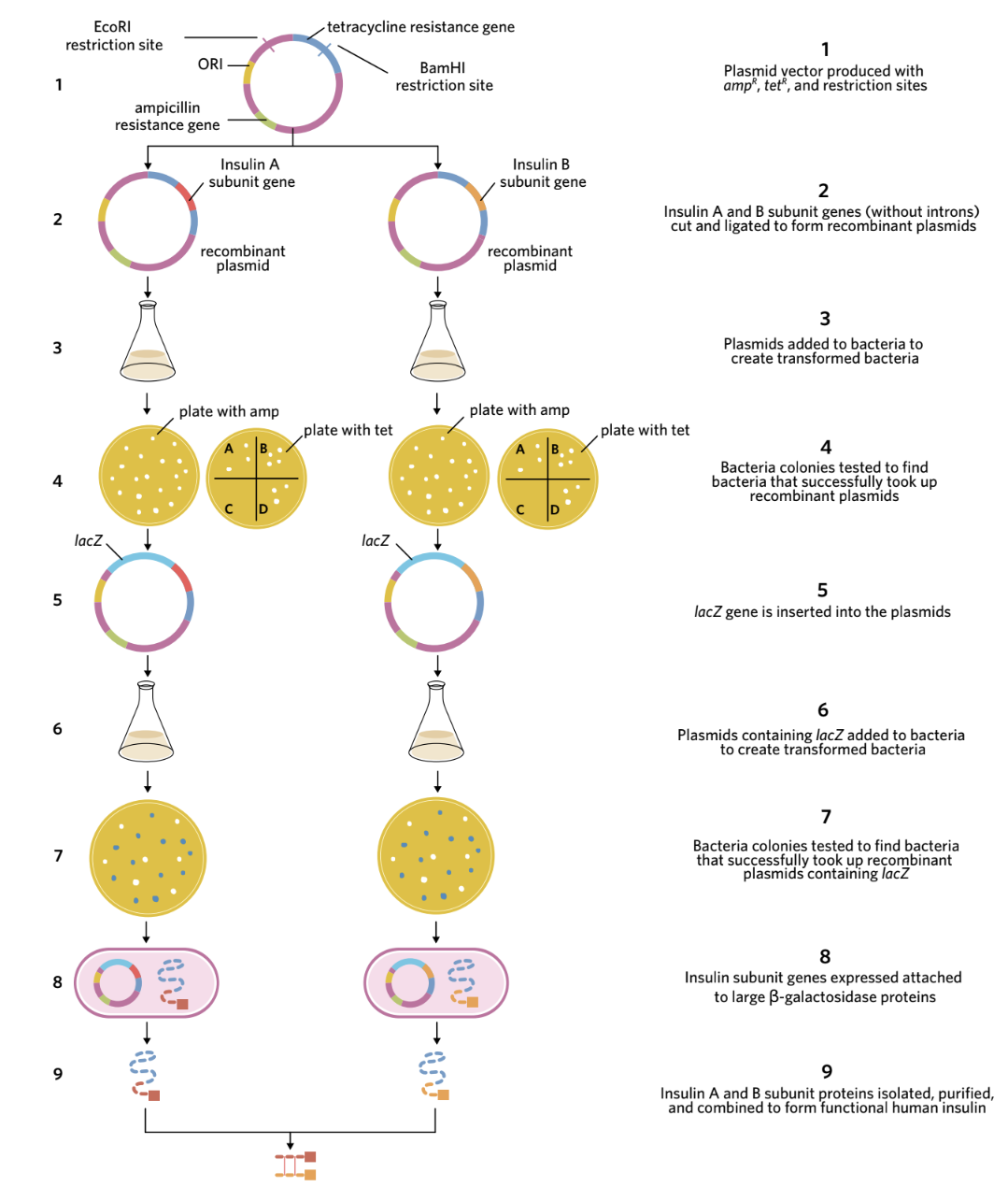
The complete process involves nine main steps:
Creating the recombinant plasmid (Steps 1-2)
Step 1: Plasmid vectors were designed containing:
- ampR gene (ampicillin resistance) - for antibiotic selection
- tetR gene (tetracycline resistance) - acting as the reporter gene
- Restriction sites for EcoRI and BamHI enzymes within the tetR gene
Step 2: Two separate plasmid vectors were prepared (one for each insulin subunit). Both plasmids, the insulin A subunit gene, and the insulin B subunit gene were cut using the restriction enzymes EcoRI and BamHI to create complementary sticky ends.
Important notes for this step:
- The insulin genes were produced without introns (made synthetically in a laboratory)
- An extra codon for the amino acid methionine was added to the start of each insulin gene
- DNA ligase joined the insulin genes to the plasmids, creating two types of recombinant plasmids
Creating transformed bacteria (Steps 3-4)
Step 3: The recombinant plasmids were added to solutions of E. coli bacteria. Some bacteria took up the plasmids through transformation.
Step 4: Antibiotic selection was performed in two stages:
First selection - Ampicillin plate:
- The bacterial mixture was spread onto agar plates containing ampicillin
- Only bacteria that took up plasmids (recombinant or non-recombinant) could survive
- Colonies that grew contained bacteria with some form of plasmid
Second selection - Tetracycline plate:
- Samples from each ampicillin-resistant colony were spread onto plates containing tetracycline
- Bacteria with non-recombinant plasmids had an intact tetR gene and survived on tetracycline
- Bacteria with recombinant plasmids had a disrupted tetR gene (interrupted by the insulin gene insertion) and could NOT survive on tetracycline
- Colonies that grew on ampicillin but NOT on tetracycline contained recombinant plasmids
- These recombinant plasmids were collected for the next step
Adding the lacZ gene (Steps 5-7)
Step 5: The recombinant plasmids were cut open again using EcoRI to insert another gene called lacZ (without its stop codon). The lacZ gene codes for ß-galactosidase, a large enzyme.
Fusion protein – a protein made when separate genes have been joined and are transcribed and translated together.
When expressed, the recombinant plasmids now produce a fusion protein - the small insulin subunit protein attached to the much larger ß-galactosidase enzyme. This fusion protein is important because the large ß-galactosidase protects the smaller insulin subunit from being destroyed by digestive enzymes inside the E. coli bacteria.
Step 6: The new recombinant plasmids (now containing lacZ) were added to fresh E. coli bacteria. Some bacteria took up these plasmids through transformation.
Step 7: Selection for bacteria with the lacZ-containing recombinant plasmids used the ß-galactosidase enzyme's function:
- The enzyme ß-galactosidase converts a compound called X-gal from colourless to blue
- Bacteria were plated on agar containing both ampicillin and X-gal
- Colonies that grew (survived ampicillin) AND turned blue (produced ß-galactosidase) contained the desired recombinant plasmids
- These bacteria could produce insulin subunits attached to ß-galactosidase
Protein production and extraction (Steps 8-9)
Step 8: The transformed bacteria containing recombinant plasmids were:
- Cultured under conditions promoting rapid reproduction
- Induced to produce the fusion proteins
- Harvested and their cell membranes broken down
- The insulin-ß-galactosidase fusion proteins were extracted
The compound cyanogen bromide was then added. This chemical breaks down methionine (which was added at the start of the insulin gene in Step 2). Breaking down the methionine separated the insulin subunit from the ß-galactosidase enzyme, allowing the insulin subunit to be isolated and purified.
Step 9: The purified alpha and beta insulin subunits were mixed together. The two chains joined via disulphide bond formation, creating functional human insulin ready for medical use.
Summary
Bacterial transformation enables scientists to produce human proteins efficiently and cost-effectively. The process involves:
-
Creating recombinant plasmids by inserting a gene of interest into a plasmid vector using restriction enzymes and DNA ligase
-
Transforming bacteria through heat shock or electroporation to introduce plasmids into bacterial cells
-
Selecting transformed bacteria using antibiotic resistance genes to identify bacteria that took up plasmids
-
Identifying recombinant plasmids using reporter genes to distinguish successful gene insertion from simple plasmid re-ligation
-
Producing and extracting proteins by culturing transformed bacteria and purifying the desired protein product
The production of human insulin demonstrates the power of this technology. By using transformed bacteria instead of animal pancreases, we can produce enough insulin for the 1.2 million Australians living with diabetes - more efficiently, more cheaply, and more ethically than ever before.
Remember!
Key Points to Remember:
- Plasmids are ideal vectors because they replicate independently from bacterial chromosomes and can be easily manipulated
- Introns must be removed from genes before insertion because bacteria cannot process them
- Four components are essential for making recombinant plasmids: gene of interest, plasmid vector, restriction endonuclease, and DNA ligase
- Plasmid vectors need specific features: restriction sites, antibiotic resistance genes, origin of replication, and reporter genes
- Two methods induce transformation: heat shock (temperature changes) and electroporation (electric current)
- Selection happens in stages: antibiotic resistance identifies transformed bacteria, reporter genes identify recombinant plasmids
- Insulin production requires TWO plasmids because insulin has two separate polypeptide chains (alpha and beta subunits) that must be produced separately then joined together