Amphibian Phylogeny (VCE SSCE Biology): Revision Notes
Amphibian Phylogeny
Introduction to phylogenetic analysis
Scientists can work out how closely different species are related using several methods. One powerful approach involves genome sequencing, where researchers compare DNA and amino acid sequences between species. By examining the similarities and differences in genetic material, we can construct phylogenetic trees that visually show potential evolutionary relationships and when species diverged from common ancestors.
This investigation focuses on using genomic data from seven amphibian species to understand their evolutionary relationships. The phylogenetic tree you create serves as a visual map of how these species are connected through evolution.
Understanding phylogenetic trees
A phylogenetic tree is a branching diagram that represents the evolutionary relationships between different species. Think of it as a family tree for organisms, showing which species share common ancestors and when they diverged during evolutionary history.
Key features of phylogenetic trees include:
- Branches: Represent different evolutionary lineages or species
- Nodes: Points where branches split, representing common ancestors
- Time axis: Usually runs horizontally, showing the progression of evolutionary time
- Distance: Species that are closer together on the tree are more closely related
The tree shows not only which species are related, but also the order in which they evolved and separated from one another. Understanding the branching pattern is crucial for interpreting evolutionary history.
Genomic data used in classification
Scientists use several types of genomic information to determine evolutionary relationships:
Genome assembly length refers to the total size of an organism's genome, measured in megabases (Mb). One megabase equals one million nucleotides. Different species can have vastly different genome sizes, which may reflect their evolutionary complexity and history.
Guanine-cytosine (GC) content measures the percentage of genome bases that are either guanine (G) or cytosine (C). Since DNA base pairing follows strict rules (G pairs with C, and adenine pairs with thymine), you can calculate thymine-adenine (AT) content using the formula:
Worked Example: Calculating AT Content
If a species has GC content, its AT content must be:
This relationship is always true because of complementary base pairing in DNA.
Protein-coding genes are sections of DNA that contain instructions for making proteins. The number of protein-coding genes can vary significantly between species and provides another data point for understanding evolutionary relationships.
The amphibian orders
Amphibians are divided into three main orders, each with distinct characteristics:
Anura includes frogs and toads. These are the most familiar amphibians, characterized by their jumping ability and lack of tails in adult form. Anurans are the most diverse amphibian order.
Gymnophiona comprises caecilians, which are limbless, worm-like amphibians. These are the least familiar amphibians to most people, often living underground or in water.
Urodela contains salamanders and newts, which retain their tails throughout life and typically have four legs.
The seven species studied
The investigation examines seven amphibian species across all three orders. Here is their genomic data:
| Scientific name | Order | Family | Genome assembly length (Mb) | GC content (%) | Protein-coding genes |
|---|---|---|---|---|---|
| Bufo bufo | Anura | Bufonidae | 5,044.74 | 44.52 | 38,109 |
| Engystomops pustulosus | Anura | Leptodactylidae | 2,555.52 | 41.97 | 48,168 |
| Microcaecilia unicolor | Gymnophiona | Caeciliidae | 4,685.94 | 44.07 | 37,109 |
| Ambystoma mexicanum | Urodela | Ambystomatidae | 28,206.9 | 40.30 | unlisted |
| Rhinatrema bivittatum | Gymnophiona | Rhinatrematidae | 5,319.24 | 44.44 | 49,282 |
| Xenopus laevis | Anura | Pipidae | 39.35 | 72,913 | |
| Xenopus tropicalis | Anura | Pipidae | 1,451.3 | 40.75 | 45,099 |
Notice the remarkable variation in genome sizes. Ambystoma mexicanum (the axolotl) has an exceptionally large genome at over 28,000 Mb, while Xenopus tropicalis has a much smaller genome at just 1,451 Mb. This nearly 20-fold difference highlights the diversity within amphibians and shows that genome size doesn't necessarily correlate with organism complexity.
Reading and interpreting phylogenetic trees
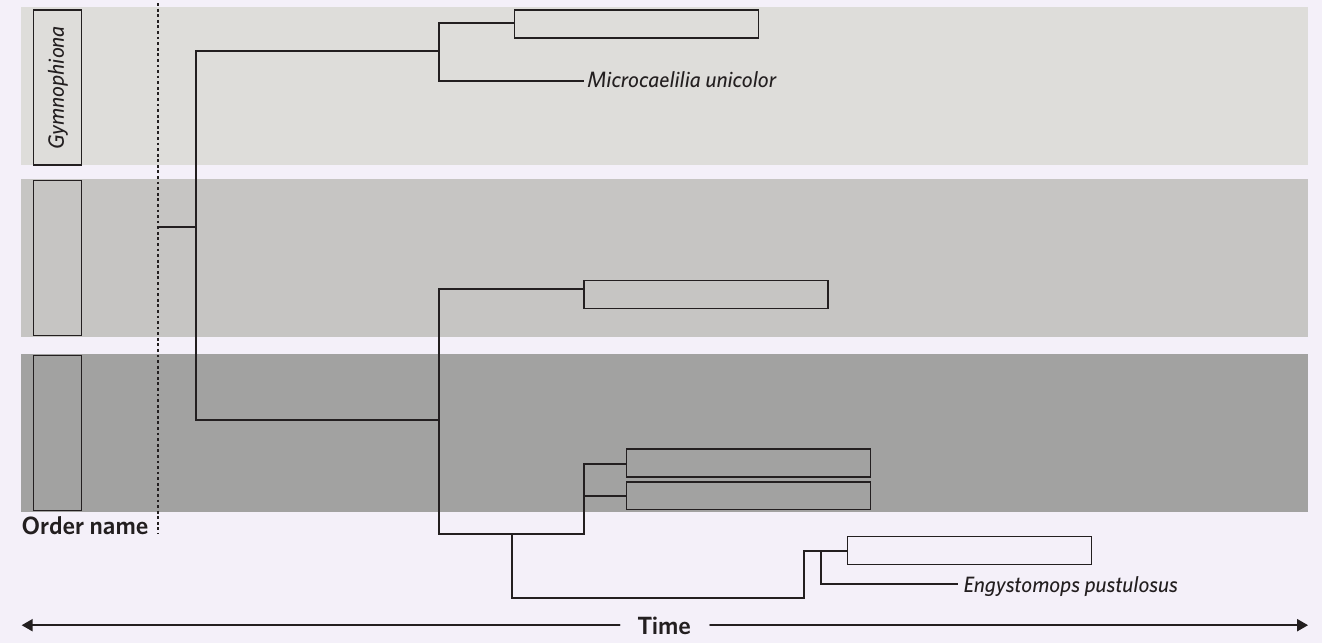
The phylogenetic tree shows evolutionary relationships through its branching pattern. Understanding how to read this tree is essential for answering questions about species relatedness.
Determining which species evolved first
Species that branch off earliest (furthest to the left on a time-based tree) evolved first. Looking at the tree structure tells you the order of evolutionary divergence.
Identifying closely related species
Species that share a more recent common ancestor (their branches meet closer to the right side of the tree) are more closely related. For example, two species in the same family will be more closely related than species from different families.
Finding ancestor-descendant relationships: In some cases, the tree may show a direct line from one species to another, indicating a potential ancestor-descendant relationship. However, most phylogenetic trees show sister species (species that share a common ancestor but neither is the direct ancestor of the other).
Comparing relatedness between groups: To determine if one group is more closely related to a second group or a third group, trace back to find the most recent common ancestor. The group requiring fewer nodes to trace back is more closely related.
Working with genomic calculations
Understanding the complementary nature of DNA base pairing allows you to calculate missing information. Since guanine always pairs with cytosine, and adenine always pairs with thymine, the percentages must balance:
- If GC content = , then AT content =
- If you need to find thymine-adenine content, simply subtract the GC content from
When working with genome assembly lengths, remember the conversion factor: 1 Mb = 1,000,000 nucleotides. This allows you to convert between megabases and individual nucleotide counts.
Worked Example: Converting Megabases to Nucleotides
Rhinatrema bivittatum has a genome assembly of 5,319.24 Mb.
To convert to nucleotides:
This equals approximately 5.3 billion nucleotides!
Key concepts for analysis
Comparing genome sizes
The largest genome in this dataset belongs to Ambystoma mexicanum at 28,206.9 Mb. This salamander has an exceptionally large genome compared to other amphibians, which is characteristic of urodeles.
Understanding AT content: To find which species has the highest thymine-adenine content, look for the species with the lowest GC content (since GC + AT = ). Xenopus laevis has the lowest GC content at , meaning it has the highest AT content at 60.65%.
Assessing taxonomic relatedness: When comparing how closely related different families are, use the phylogenetic tree structure. Families within the same order will be more closely related to each other than to families in different orders.
Exam tips
When answering phylogeny questions in exams:
- Always refer to the tree structure when determining relatedness
- Show your working when calculating nucleotide content or genome sizes
- Remember that closer branching points indicate more recent common ancestors
- Use the time axis to determine which species evolved first
- Consider all three orders of amphibians when making comparisons
Remember!
Key Points to Remember:
- Phylogenetic trees are visual diagrams showing evolutionary relationships between species, with branches representing different lineages and nodes showing common ancestors.
- Three amphibian orders exist: Anura (frogs and toads), Gymnophiona (caecilians), and Urodela (salamanders and newts).
- GC content and AT content are complementary - they always add up to because of DNA base-pairing rules.
- Genome size varies dramatically between species - in this dataset, from 1,451 Mb to 28,206.9 Mb - and doesn't necessarily correlate with evolutionary complexity.
- Closer relationships on phylogenetic trees are shown by more recent common ancestors (branching points closer to the present on the time axis).